关键词
结构 序列 算法 分辨率 建模 人工智能 图像处理 深度学习
会议回顾
2024年5月17日,广州实验室苗智超研究员邀请了华中科技大学的黄胜友教授进行了线下及线上转播的学术报告“基于人工智能的冷冻电镜RNA三维结构建模”。黄胜友教授长期从事计算生物物理和生物信息学方面的研究,在Nature Protocols, Proceedings of the National Academy of Sciences,Nucleic Acids Research, Bioinformatics,Drug Discovery Today,Physical Review Letters等国际一流期刊上发表SCI论文80余篇,其中第一和通讯作者50余篇,文章累计他引2000余次。所提出的集合分子对接算法和迭代打分函数模型被国际同行广泛采用,发展的蛋白质分子对接算法和打分函数模型在称为国际蛋白质相互作用预测的“奥林匹克竞赛”CAPRI评估中多次排名第一。
会议内容
继中心法则发现以后,我们认为DNA是遗传物质,蛋白质是生命活动的执行者,对RNA有所理解过后,再返回看中心法则,会发现RNA才位于生命活动中承上启下的中心位置。实际上,从某种意义上来说,RNA也是最复杂的。因为蛋白质的结构是相对比较稳定的,当然这也是Alphafold可以一定程度将蛋白质结构预测的很好的重要原因。
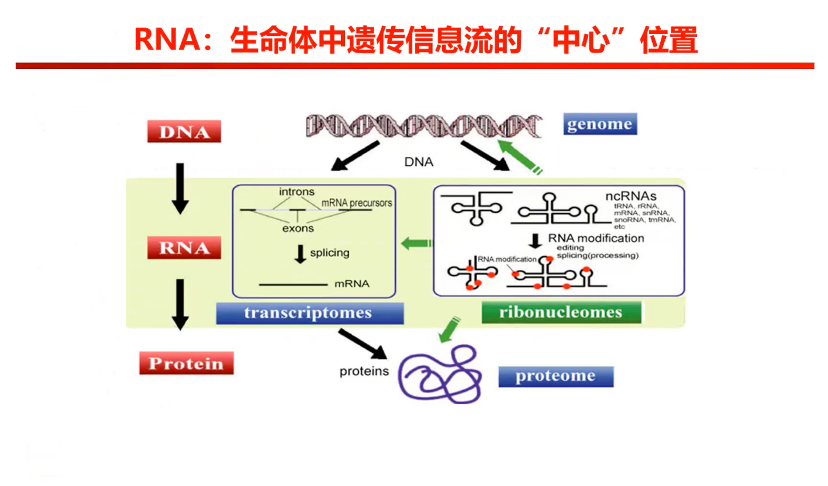
图1. RNA处于生命体遗传信息流的中心
DNA的双螺旋结构相对RNA来说也是是非常稳定的。RNA既存在transcriptomes也存在ribonucleomes,情况相对更复杂,也是我认为Alphafold在RNA结构预测上表现欠佳的可能原因。RNA的三维结构要怎么确定呢,一般来说做实验得到的三维结构更准,但往往也伴随成本高难度大的问题,相对来说,通过计算得到三维结构的成本更低,速度或周期更短。在此基础上,我们进一步对RNA三维结构预测问题进行简化,就是在给定的一段RNA序列,要怎么得到它的三维结构。
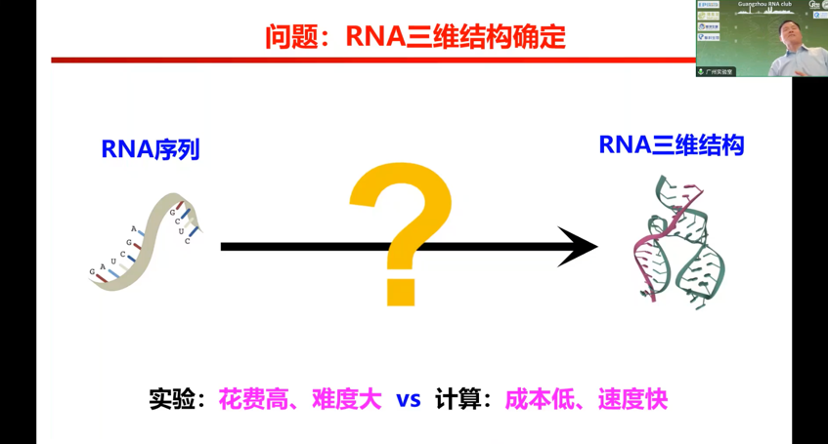
图2. RNA三维结构要如何确定?
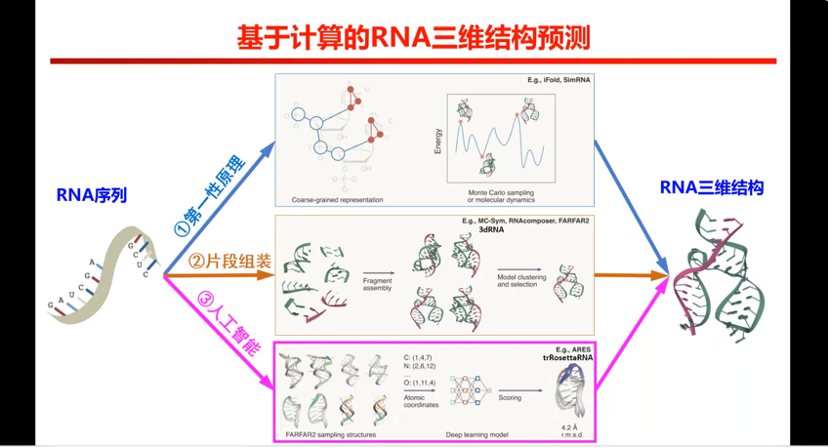
图3. 计算预测三维结构的三类方法
目前来说,从计算上来说有三类方法来预测RNA三维结构。第一类方法叫第一性原理,RNA碱基一共四种(A,U,G,C),根据序列上碱基的顺序进行拼接折叠即可。但是这种情况的话它自由度太大,如果我们一个原子的话有三个自由度,那么几千个原子就相当于就是几万个自由度,想要正确的结构是很困难的。所以为了解决这个问题,后面就提出了第二种方法,这种情况叫片段组装。我们就依据序列把它简化成片段的形式。举例来说,我把序列分成 10 个片段,那么每个片段的话自由度只有3,那我们只有 30 个自由度,计算数量级就急速地下降。
目前来说,片段组装的话是现在最主流的预测方法,包括国外的RNAComposer,还有FARFAR都属于这方面。国内肖毅老师的3dRNA也是这方面的一个工作。当然方法也有局限性,把RNA拆成片段,如果没有生成没有结构里面需要的片段,就无法拼出正确的三维结构。
那么是不是存在一种方式是万能的,预测的各种情况都可以搞定。理论上是大家还是很期待人工智能在预测方面的表现。近期关于人工智能在预测方面也有几个工作发表,一个是ARES,是斯坦福的工作;trRosettaRNA是山东大学杨建益老师的工作,还有最近的RosettaFoldNA,以及最新的AlphaFold3。可能是因为Alphafold把大部分蛋白质结构解决得很好,好像RNA预测问题也可以借助于AI解决。实际上,现在人工智能属于数据驱动的,所以某种意义上,没有数据,人工智能什么也做不到。
所以如果现在没有一种方法能够解决RNA结构预测问题,那么用多模态策略,使用多种方法,把所有的数据都用上,一个信息不够,那么用两个、三个、四个,十个指不定就解决了。所以出现了一个概念,叫做整合策略,或者叫整合建模。
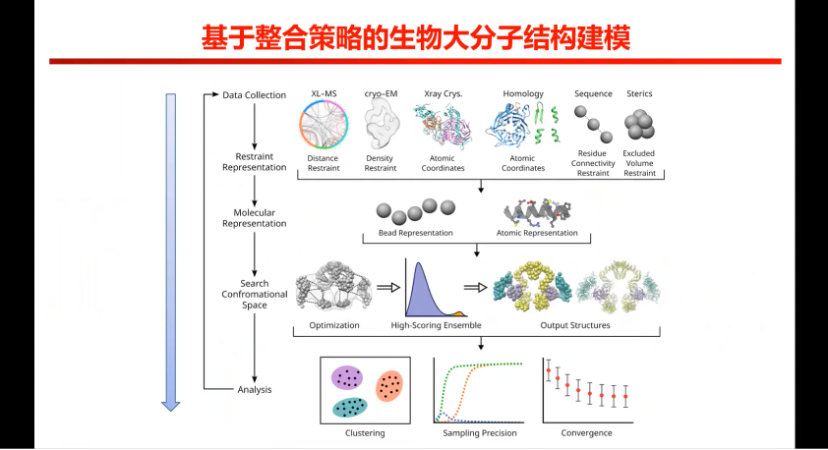
图4. 生物大分子结构建模
一种数据的话只能看到一个方面,整合多种数据就可以得到多个方面信息。第一步,整合实验信息,例如Xray,cryo-EM等;第二步分子表征这些信息,得到通过实验的到的结构方面的约束信息;第三步,采样所需要的结构信息;第四步,筛选出正确的采样策略。实际上现在的AlphaFold3的扩散模型就是这种模式,扩散模型可以挑选出很多预测结构,中间过程设置打分函数挑选出正确的预测结构。这应该也是后续结构建模的一种趋势。
我们聚焦于冷冻电镜方面也是因为我们有比较多这方面的实验信息。在做的过程中发现冷冻电镜信息有一个非常明显的特点,就是它实际上是个高质量数据和低质量数据的完美结合。
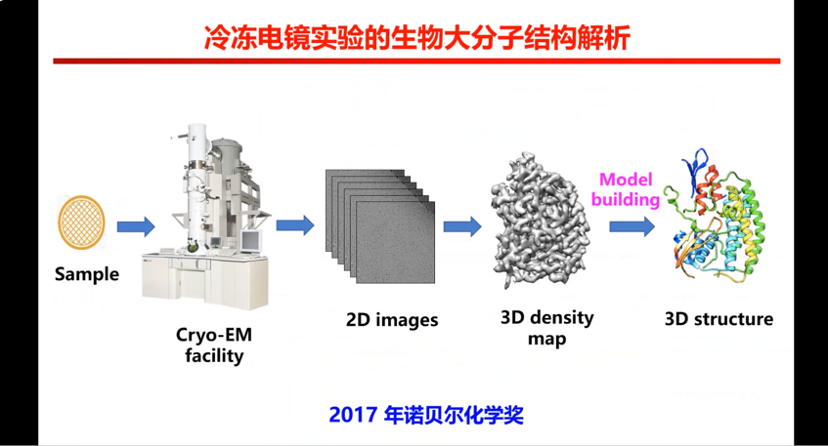
图5. 冷冻电镜帮助生物大分子结构解析
举例来说,冷冻电镜做得好分辨率可以很高,做得不好也可能很不好;第二,它同一个结构分辨率好的地方可以达到一点几埃,不好的地方可以达到二十几埃,所以某种意义上也是我们的机会。
我们主要是集中2D图像生成3D density map。就像我们前面说的一样,这个冷冻电镜数据的话是真是高质量和低质量完美结合,它有3埃的时候,原子细节都可以看的比较清楚。比 3埃差的话,差不多5埃就基本上都看不到原子了。
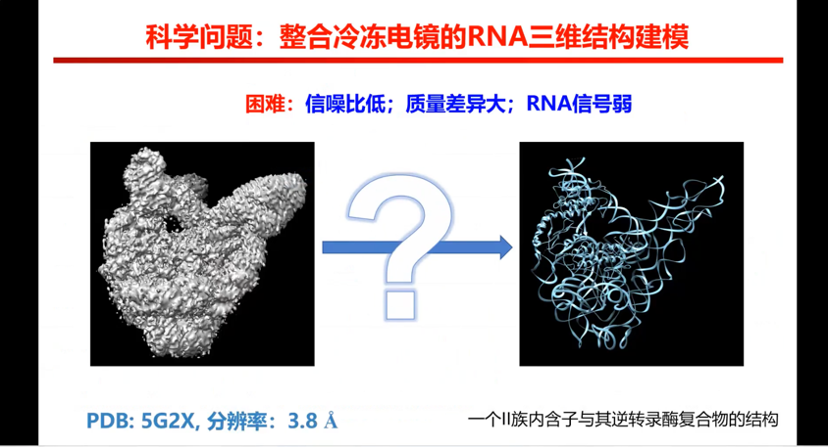
图6. 整合冷冻电镜数据进行RNA三维结构建模存在的问题
既然有很多冷冻电镜数据这么难解,我们是不是就可以来做这个事情?以上述图例来说,大家会发现里面信号信噪比很低,质量差距特别大,RNA的信号也比较弱。第一眼看不出来怎么得到右边的RNA结构。所以我们就做了一个人工智能的框架,通过人工智能,提取右边的结构信息。
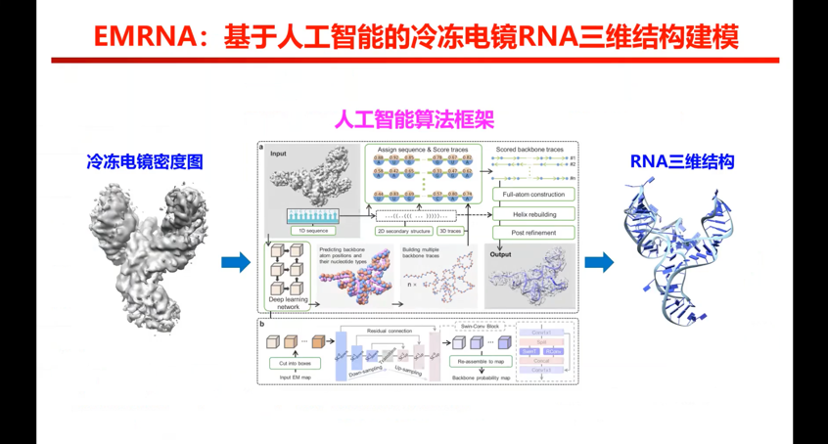
图7. EMRNA基于人工智能的冷冻电镜RNA三维结构建模算法概览
大概流程如下,流程图最左边是冷冻电镜的密度图,右边是结构,我们在中间提出了一个自动化的人工智能的算法框架(EMRNA) , 相当于你给它一个密度图的话,它就给你一个结构。模型核心划成几个模块,第一个步骤相当于RNN 主链原子信号的挖掘;第二步要把它连起来并成链,但主链是有位置没有序列,因此我们要把序列信息添加上去;就是第三步结合二级结构和序列信息,把序列装上去。
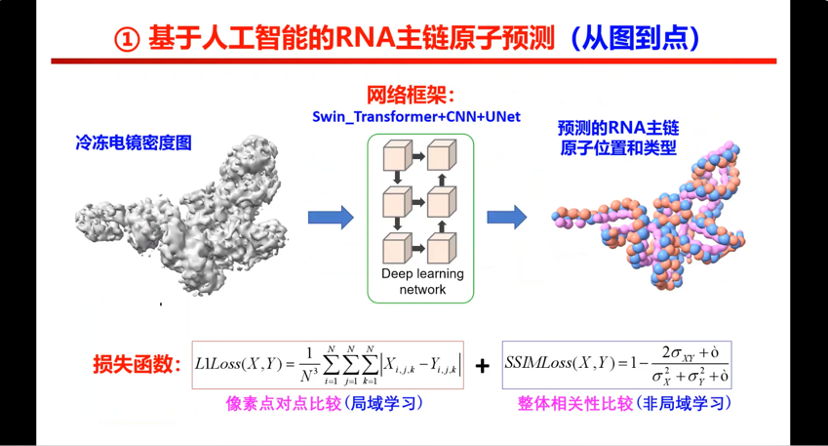
图8. EMRNA主链原子预测概览
主链原子的挖掘或者预测,我们用简单的四个字描述就是从图到点。我们这个算法能够做到准确地从那些看起来很模糊,好像都没有信息的部分,把原子信息挖掘出来,最核心的就是这个网络。我们做的好有两个方面原因,第一个方面,网络框架很特殊,一个是Transformer,一个是CNN,一个是UNet,三个模块功能的综合应用,可以做到又快又好,在做好短程碱基间作用的同时兼顾RNA远距离相互作用的场景,同时提高算法的整体速度。第二个方面,核心损失函数的创新。我们也用了两个模块,第一个做叫 L1Loss,是负责局部学习的;第二个叫SSIMLoss,负责整体相关性比较。在保证点对点信息还原的同时,兼顾对预测结构整体性的考虑。
这从图到点的过程相当于我们获取核苷酸的位置,那么只有位置是不够的,我们要把它串起来。
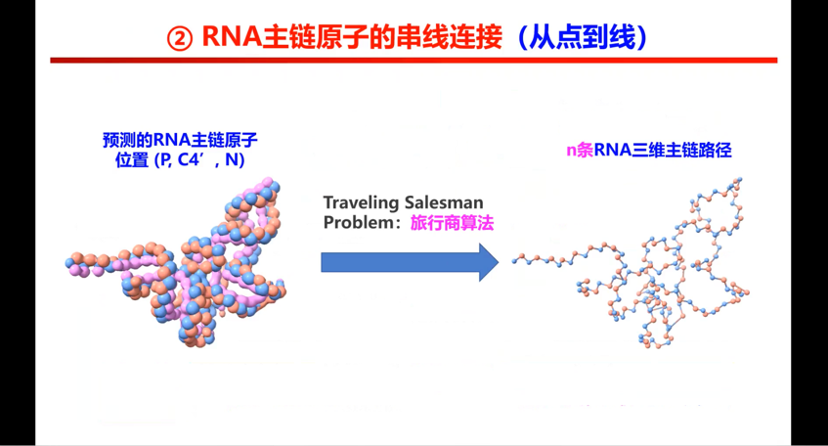
图9. EMRNA主链原子的串线连接
核苷酸串起来过程我们称之为从点到线,使用了旅行商的算法。因为其实RNA的实际信号是比较弱的,所以某种意义上说,得到多条,上万条都有可能的,我们需要依据序列信息去判断哪条是对的。
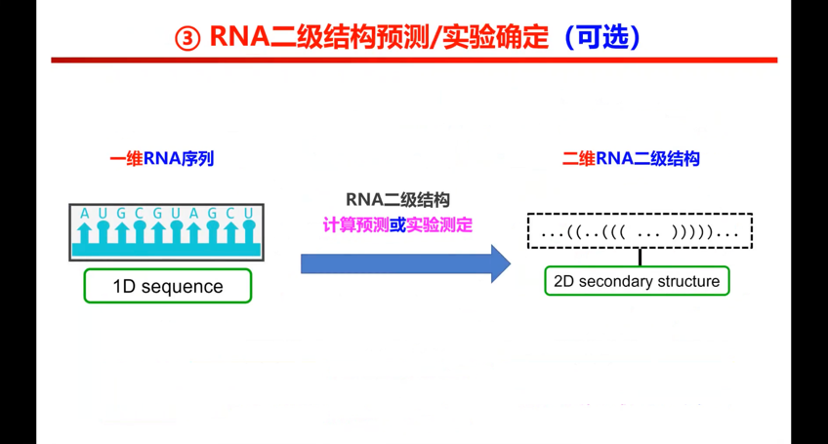
图10. EMRNA中RNA二级结构确定
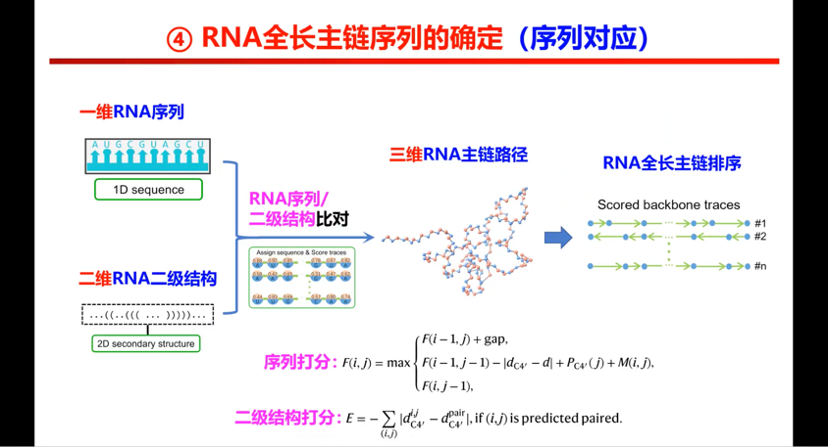
图11. EMRNA中RNA全长主链序列的确定
增加序列部分的信息需要一个二级结构去帮它去打分。为什么用这个二级结构或加一个二级结构的预测呢?主要是二级结构的预测比较自动化且比较准确,用我们之前已知的一维的序列,还有预测的二级结构这两个信息,装在从图像中捕捉的主链。怎么把序列装对这个事情,可以参考现有的一些算法。
序列比对或者结构比对的问题,就相当于怎么样把一维的序列和这二维的结构信息匹配到那个三维图像上。这实际上是个动态规划的事情,相当于这里面包含两个打分,一个序列打分,一个二级结构打分。用这两个打分作为评估,进行主链装上序列信息的训练。每一条主链装上的序列后,它的得分是不一样的,我们就挑第一名。基本上第一名如果不对,其他的对的可能性也不是太大。
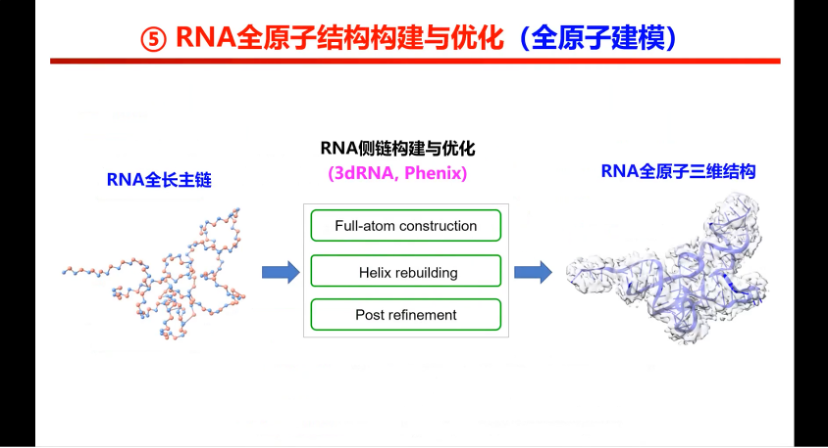
图12. EMRNA中RNA全原子结构构建与优化
到这个地方为止,主链位置也好了,序列信息也好了。最后一步,已经有RNA三维结构的backbone, 主链定了,序列核苷酸信息确定以后,就可以直接构建全原子的三维结构。这一步我们没有创新的算法在里面,用已发表的算法,例如Phenix或者3dRNA都可以搭建全原子模型。
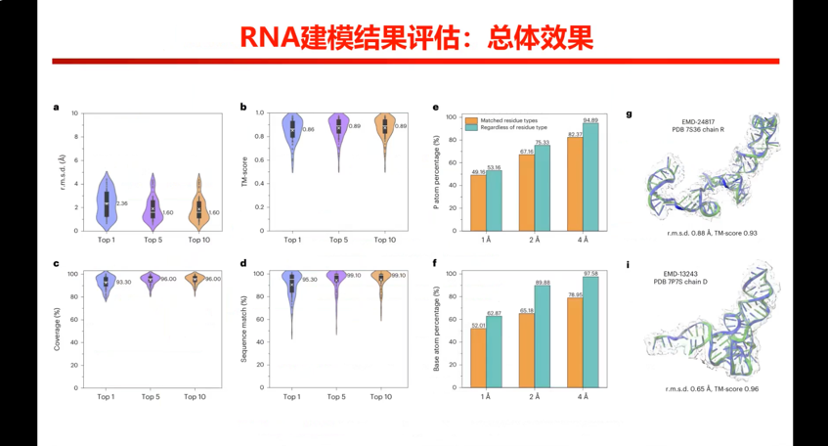
图13. EMRNA中RNA建模结果评估
所以整个流程就是这样,最核心的就是我们前面那个网络框架,怎么从那个很模糊的粒度里面能够准确地定位主链原子位置,后面都是工程学的事情。算法表现方面,我们做的评估图例比较多,RNA结构方面,一个是用RMSD作为评估标准,相当于比较预测结构和PDB真实结构相差有多远。
第二个标准叫做TM score,可能RNA方面做Cryo-EM,TM score 不一定适合,但TM score用来比较与真实PDB结构一致性程度,一致性越高越好。第三个是coverage,用过Phenix 都知道以案例有一千个碱基来说,软件可以构建出多少碱基;第四个是序列正确率,sequence match,装序列装得对不对?也是越高越好,除了第一个标准是越低越好之外,其他都是越高越好。最右面的图,一个蓝色的,一个绿色的,一个是解析PDB 结构,一个是预测的RNA结构,总体效果还是不错的。
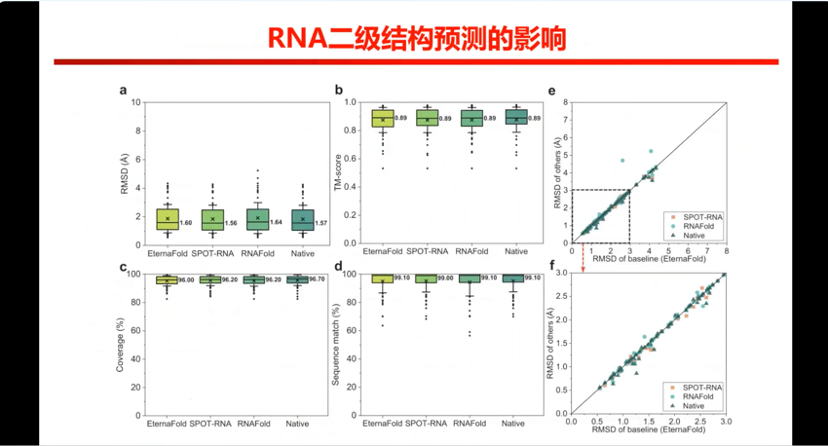
图14. EMRNA中RNA二级结构预测的影响
接下来我们看一下,二级结构的影响,因为我们用二级结构去打分,作为一个评分标准。图例上一种颜色代表一种二级结构的准确度,基本上差别不是太大。
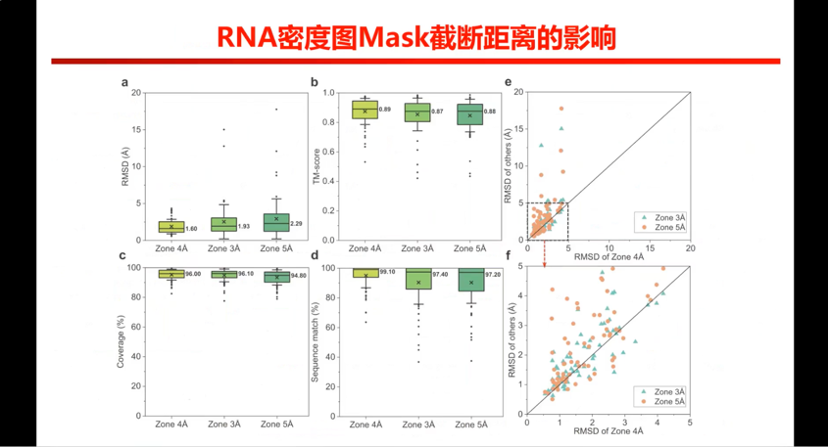
图15. EMRNA中RNA密度图Mask截断距离的影响
第三个是 RNA 密度图Mask截断距离的影响,为什么做这个是因为很多结构是个蛋白和RNA的复合物,所以预测要先把 RNA mask 出来。当时存在一个疑问,是mask的大小存在多大的影响,所以我们就做了一下mask在3埃、4埃、 5埃都做了一下,差异不大。每种颜色都代表一种mask的影响,但是mask还是对技术有要求,如果mask 不好,它可能不一定能做出正确的结构。
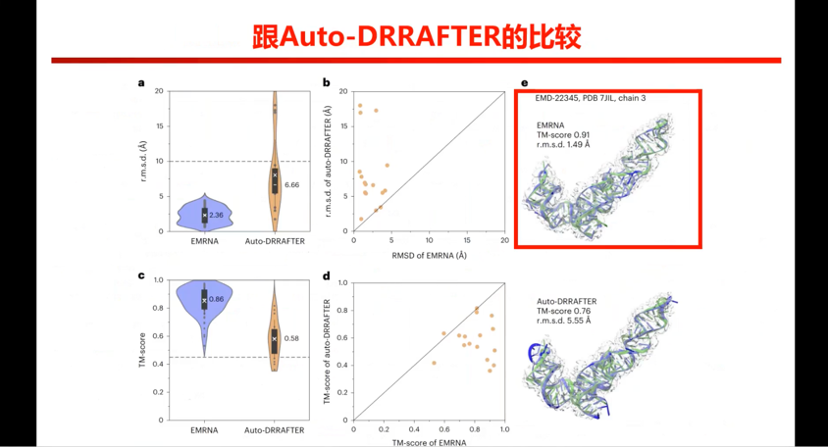
图16. EMRNA与Auto-DRRAFTER效果的比较
首先就是与Auto-DRRAFTER的比较,这个是斯坦福那边做的,他们应该是除EMRNA之外,他们唯一一个能够做全长的。左边这个是我们的EMRNA,右边是Auto-DRRAFTER的,我们比他们好不少。同时EMRNA还有一个优势,就是比较快,因为他们估计一周做一个100个核苷酸的,EMRNA使用一个GPU 估计几分钟就好了。
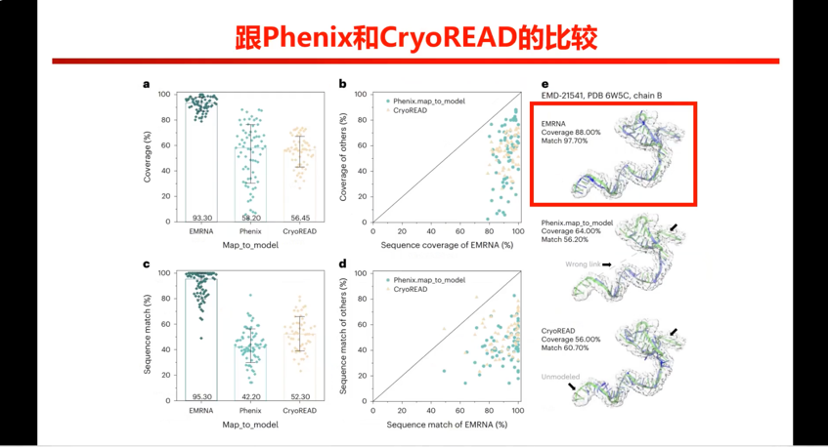
图17. EMRNA与Phenix以及CryoREAD的比较
还要与领域里面表现比较好的算法都进行比较,例如Phenix还有CryoREAD(去年12月份发在nature methods)。结果部分,中间是 Phenix ,右边是CryoREAD,EMRNA都好不少。
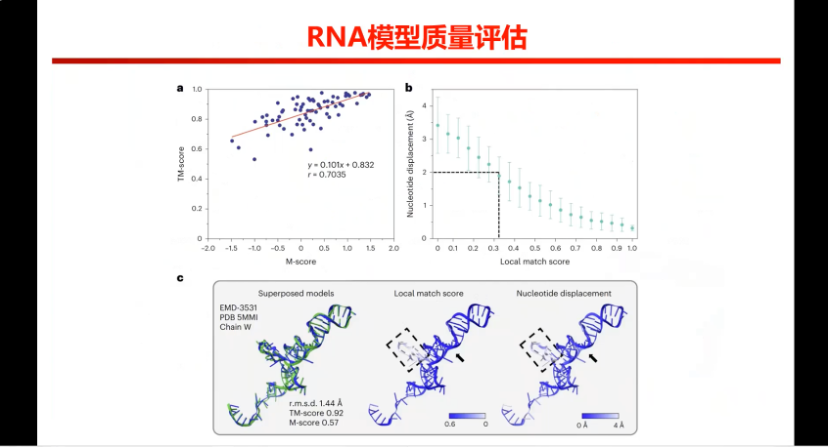
图18. EMRNA产生的RNA模型质量评估
结构建模里面还有一个非常重要一个方面,就是预测结构的可靠性。所以进行了RNA 模型的质量评估,我们专门设计一个参数叫M-score,M-score和结构的 TM-score就是预测的结构,好不好的标准。两个score之间也有很好的相关性,所以M-score可以告诉实验学家这个结构到底好不好。

图19. 其他工作之EM2NA
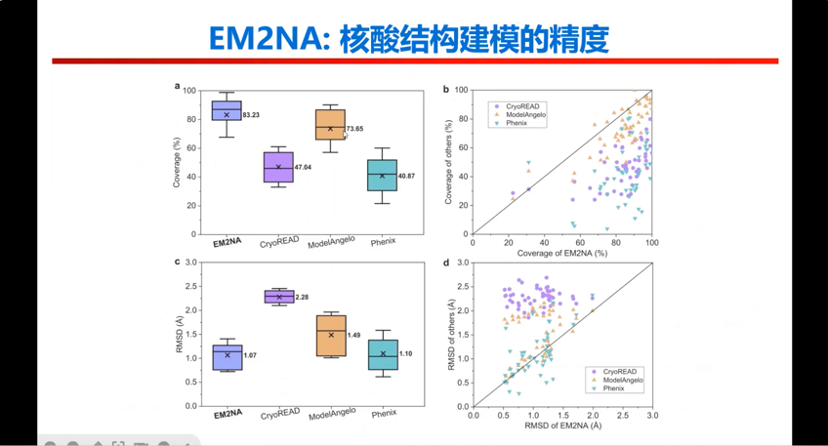
图20. 其他工作之EM2NA精度比较
我们想再跟大家分享两个相关的工作,一个叫做 EM2NA,相当于EMRNA的一个 2.0 版本。因为前面只能够做RNA,但是还有很多DNA的结构。所以我们把它做成一个统一框架,不管 RNA或DNA都可以做,所以叫做EM2NA。框架和EMRNA差不多,功能上既能够处理DNA,也能够处理RNA,能够自动地从那个密度图里面探测DNA来帮你建模,也可以直接直接从那个准确的模型进行建模。精度层面,EM2NA与CryoREAD, ModelAngle, 还有Phenix,比较的结果都是EM2NA还不错。
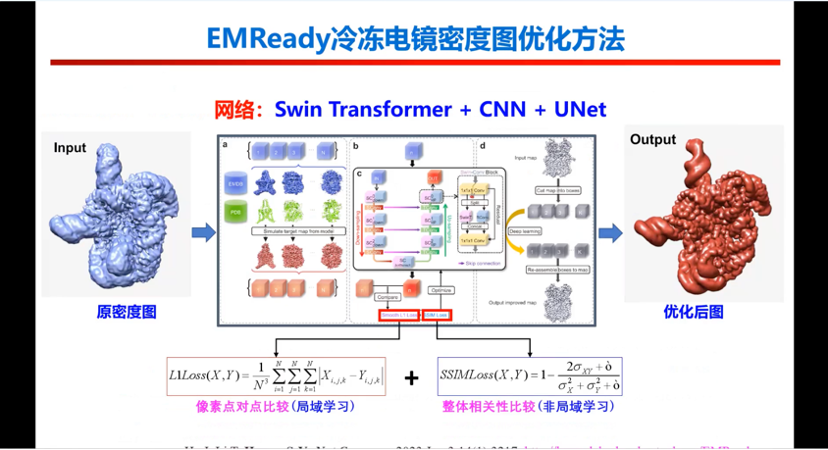
图21. 其他工作之EMReady
第二个我们想分享的工作是EMReady。实际上是针对于自动优化那个冷冻电镜密度图的工作。
这个工作比较好的地方是我们可以把这个密度图优化得挺好。左边是个蛋白和RNA的一个复合物,这个信号实际上是很模糊的,经过我们模型网络优化以后,RNA的双螺旋会变得非常清楚,如果分辨率可以更好一点,它可以把碱基配对的看得非常清楚,可以看到有原子细节在里面。
如果大家不需要一步到位获得电镜解析的RNA结构,可以用这个模型优化冷冻jian j就可以了,这个网络框架实际上和我们前面基本上是一样的。我们这个工作方法能力更好,鲁棒性更好。
精彩问答
问题1:您这个主要工作是对 DNA以及RNA的结构优化,那么如果是一些蛋白质的结构,您这个模型能不能也进行优化?
答:我们这个算法实际上对DNA/RNA/蛋白都是可以做的。实际上的话,我们EMReady就是专门优化密度图的,就是刚开始说CryoEM实验很模糊或者看不到原子,我们可以优化以后蛋白和 RNA/DNA都可以同时优化得挺好。拿到这个优化好的图去解结构,就会感觉比较容易。
问题2: 如果是有一个大概 4A 的 RNA结构,您这个 EMRNA 算出来的它的准确率大概是多少。
答:这个问题很好,这种情况的话, 4A的话,它可以从两个方面来看它的准确率多好,一是你的mask的水平,你mask不好的话它可能准确率没那么高;如果你用通用的序列准确率估计,如果你 mask 好,它序列准确率可以达100%。mask 不好的序列准确率大概只有 50%~60%左右。第二个,如果你从准确率来说,你只是说我那个主链有,不管它序列的话,不管它侧链的话,那么它的准确率是挺高的,应该百分之八九十,问题不大。所以这两个问题,第一个是你是不是把那个主链建好了?这个准确率在百分之八九十没有问题。如果你是说是不是把那序列组装对了,这个里面就因你的 mask水平,还有整个的分辨率有关。
黄胜友教授此次会议报告已收录于Guangzhou RNA club bilibili视频网站(https://www.bilibili.com/video/BV1js421w7mp/?spm_id_from=333.999.0.0)
欢迎关注Guangzhou RNA club公众号、网站(rnaclub.rnacentre.org)、twitter(@RNA_club)。
 Guangzhou RNA club
Guangzhou RNA club