会议回顾
2023年5月22日,中山大学中山医学院王金凯教授邀请了邢毅教授进行了线上学术报告“Long-read strategies to study the human transcriptome”。邢毅教授是费城儿童医院(CHOP)Francis West Lewis讲席教授,计算和基因组医学中心主任,生物医学和健康信息学系主任,以及宾夕法尼亚大学(Penn)病理学和实验室医学系的教授。邢毅教授的实验室专注于RNA相关的计算生物学和基因组学,以及它们在人类遗传学和医学中的应用。
讲座内容
邢毅教授的研究针对于RNA表达调控相关的生物信息学和基因组学,他们把这些发展的新技术用在研究RNA在人类疾病和遗传学上的作用。他们实验室近几年比较感兴趣的一个方向是怎么用新的长读长RNA测序技术来研究人类的转录组。
邢毅教授实验室最感兴趣的一个方向是RNA可变剪切,RNA可变剪切的方式是多种多样的,有外显子跳跃、外显子互斥、内含子保留等。有很多比较长的人类基因有很多个外显子,然后它会产生比较复杂的可变剪切方式,通过排列组合,有些基因可能产生成千上百的可变剪切产物。有很多人类疾病是通过基因组里的突变影响了剪切调控,然后产生了异常的可变剪切异构体,使RNA或者蛋白质的功能失调,最后导致了人类的疾病(图1)。
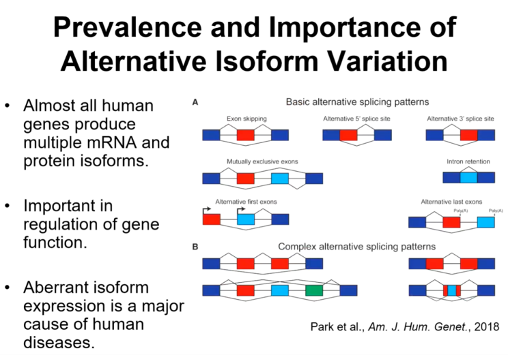
图1. RNA可变剪切
在过去的大概15年内,大家用来研究可变剪切或者研究转录组最常用的一个高通量手段是通过RNA测序。
但是这种传统的短读长RNA测序有一个最大的问题,就是它不能让你读到全长的转录本。在这里,邢毅教授给大家举了一个很简单的例子。有一个比较长的人类基因,它有非常多的外显子以及可变剪切的方式。传统的RNA测序实际上是要先把这种全长的RNA打断成一个个小的片段,然后测这些小的片段。所以我们最后得到的测序数据不是全长的转录本,而是一个个小片段。但是很多情况下我们想知道的是最后产生的这种全长的转录本和它对应的蛋白产物是什么样的。我们通过这些小片段测序数据很难知道到底是第一个转录本存在还是第二个转录本存在,以及它们的表达量是多少(图2)。
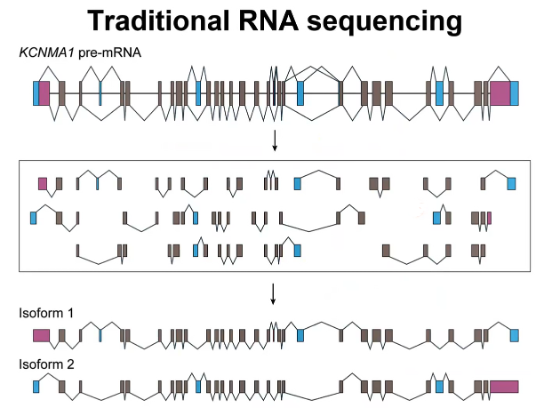
图2. 传统RNA测序
因此在过去的十年内大家对新一代基于长读长的测序平台有很大的兴趣,
现在就是大家用的比较多的两个平台一个是Pacific Biosciences(PacBio),另外一个是Oxford Nanopore Technologies(ONT)。Oxford Nanopore Technologies基本的工作原理是它通过一个马达蛋白把DNA或者RNA分子拽过一个纳米孔,然后每一次有一个碱基经过这个纳米孔时,它会产生一个电流扰动,所以最后读到的是电流数据,然后我们通过这个电流数据可以推测是什么样的碱基在经过这个纳米孔。这个技术一个很好的地方是它可以轻松读到特别长的序列,如超过1万个碱基对的序列。它基本上能够覆盖整个人类转录组里所有的转录本长度,因为大多数人类转录本平均长度大概是在1-2 KB,有一些可能比较长到10 KB。
所以这种长读长测序平台能够让我们读转录本时从一端读到另一端(图3)。
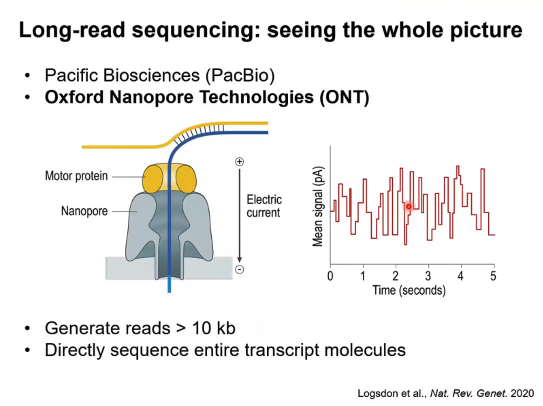
图3. 长读长测序
于是邢毅教授开始考虑长读长测序的应用场景,
他们的第一个计划是用长读长测序来测序一类比较特殊的RNA,叫做环形RNA。我们知道大多数的真核生物里面的RNA在剪切的时候是以线性的方式来剪切的,就是从前往后走,最后得到这种经典的线性RNA产物。但是在一些情况下剪切可以有非线性的方式,pre-mRNA通过一个叫backsplicing的下游的剪切位点攻击上游的剪切位点的反应,最后形成一个环形的RNA。环形RNA大概在十几年前通过短读长测序被发现是非常普遍存在的,它也有各种各样的生物学功能。但是有一个问题是鉴定全长的环形RNA是一个很难的事情,比如说外显子4回到外显子2,当你用短读长做测序的时候,你会有一些测序片段掉在外显子2、3、4里面,但这种情况下很难判断这些片段到底是来自线性RNA还是来自环形RNA。所以邢毅教授他们就想能不能开发一个对环形RNA进行长读长测序的工具(图4)。
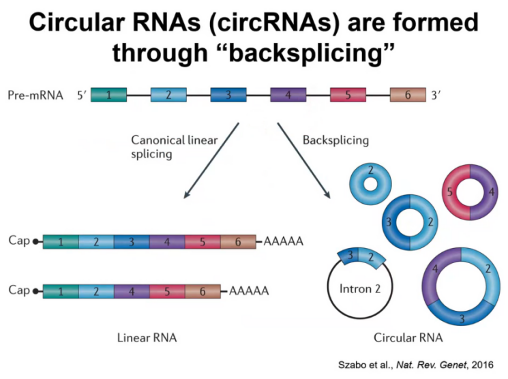
图4. 环形RNA通过backsplicing形成
这个工作是邢毅教授实验室两年前发表的,
他们做了一个叫做isoCirc的工具,其可以通过长读长的纳米孔测序来读全长的环形RNA。基本的流程如下,首先从样本里面把RNA提取出来,去掉线性RNA和rRNA,接下来通过随机引物对环形RNA做反转录,当完成反转录之后用连接酶填成环形DNA模板,然后最重要的一步是他们做了一个叫做滚环扩增的反应。这样通过一个特定的模板可以产生特别长的滚环扩增产物,这个滚环扩增产物是同一个模板重复多次得到的。得到滚环扩增产物后用纳米孔进行测序,对于得到的测序数据,他们发展了一系列的计算流程,从而得到高置信度的环形RNA异构体。当时邢毅教授实验室用isoCirc分析了大概十几个人类的组织,然后得到了超过10万个环形RNA异构体,其中有一些环形RNA的异构体非常长,没有办法用简单的方式获得(图5)。
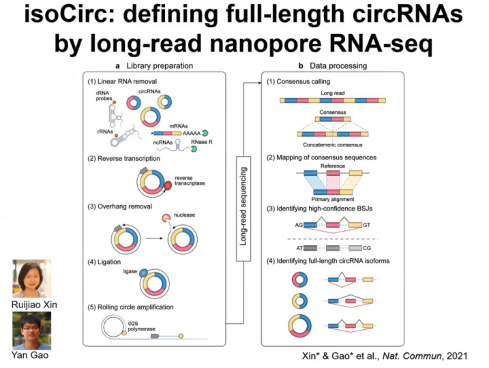
图5. isoCirc:通过长读长纳米孔RNA测序读出全长环形RNA
长读长RNA测序在过去的十年内随着它技术平台的进一步的发展得到了更多的关注,可以看到相关文章数量在逐步上升。然而把长读长RNA测序和短读长RNA测序放在一起来看的话,可以发现在整个基因组领域短读长的RNA测序现在仍然是绝大多数人使用的工具(图6)。这就产生了一个很有意思的问题
到底是什么阻碍了人们选用长读长RNA测序?
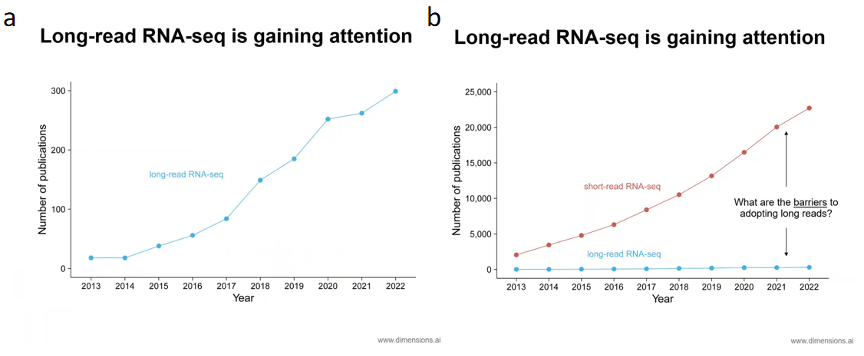
图6. 长读长RNA测序得到的关注
长读长测序平台有两个比较大的缺陷,
一个是它的测序吞吐量要低一些,还有一个是长读长测序的错误率会比较高。邢毅教授向我们展示了他们实验室在人类血细胞细胞系上产生的Oxford Nanopore cDNA测序数据,这是肿瘤免疫中的一个明星分子CD19。每一条线都是一个RNA测序序列,它们确实是覆盖了整个转录本,且有外显子的地方就有信号,没有外显子的地方就没有信号。但是如果我们仔细关注到某一个特定区域的话,会发现它有非常多的错误,这些错误最后会影响到我们对剪切位点的鉴定或者是对全长转录本的发现。因此在将近十年之内,大家觉得比较靠谱的技术路线是不能只看长读长的RNA测序,而是需要在同一个样本上产生一个短读长的RNA测序,然后用这个短读长的RNA测序数据来校正长读长RNA测序的结果。
所以邢毅教授感兴趣的是能不能只用长读长的RNA测序去比较准确的推测剪切位点或者转录本异构体(图7)。
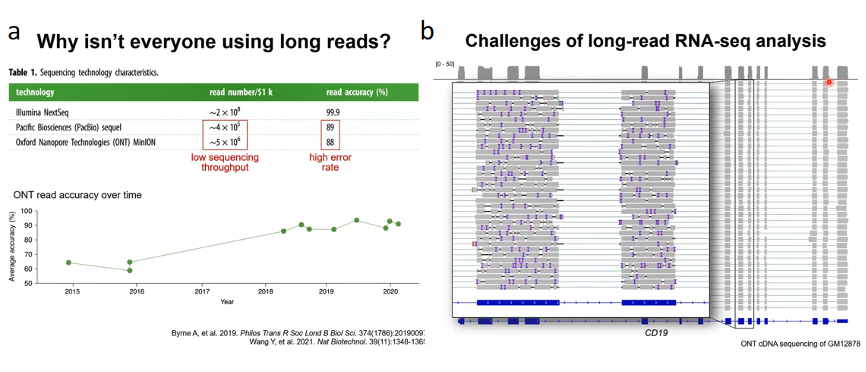
图7. 长读长RNA测序的缺陷
邢毅教授实验室做的第一件事情是用了一个合成的叫synthetic spike-in的RNA,它总共有68个RNA。这68个RNA已知外显子和内含子的结构及每个RNA的浓度。接下来,他们对这些RNA进行测序,然后寻找能区分正确剪切位点和错误剪切位点的特征。他们发现如果在剪切位点处10bp的碱基没有任何错误,其剪切位点的错误率不到2%。但是如果在剪切位点处10bp的碱基存在错误,其剪切位点的错误率能达到10%。因此,若
在剪切位点处获得完美比对,其剪切位点的可信度能得到提高,这是其中一个特征。另外一个是如果
一个剪切位点在不同的reads里面反复出现的话,这样的剪切位点也比较可靠(图8)。
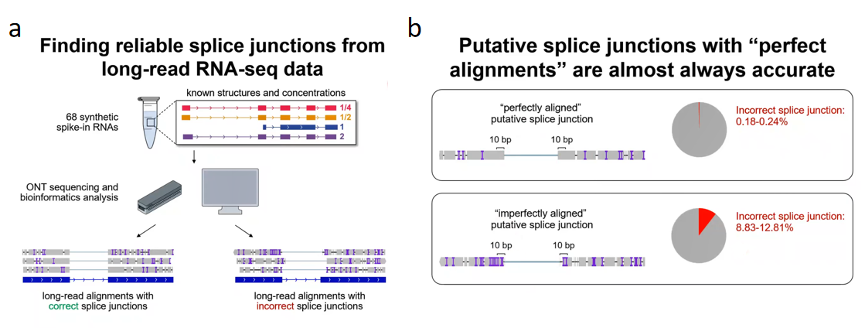
图8. 区分剪切位点正确与否的特征
基于以上两种特征,邢毅教授实验室发表了一个新的
做转录本发现和定量的工具叫做ESPRESSO。ESPRESSO主要原理如下,如果有一个经典物种的基因去做长读长RNA测序,首先我们可以获得该物种的参考转录本注释。对于一个特定基因而言,它有不同的reads,可能有一些reads的某些地方有完美比对剪切位点,另一些地方有不完美比对剪切位点。我们拿这些长读长测序的比对结果加上参考转录本注释,可以得到我们认为有高置信度的剪切位点,即完美比对剪切位点。这些剪切位点可能是我们已知的剪切位点,也可能是新发现的剪切位点。如果这些新发现的剪切位点在2-3条reads上是完美比对的,我们也认为这些新发现的剪切位点是高置信度的(图9)。
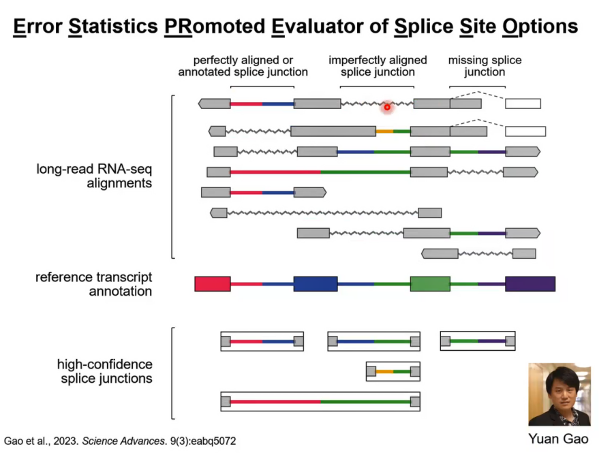
图9. ESPRESSO原理1
把这些剪切位点都拿出来以后,对每一条long-read的数据进行校正。比如说这一条long-read在第二个剪切位点上是不完美比对的,但这个不完美比对剪切位点和两个高置信度剪切位点有重叠,所以我们想确定这里的剪切位点到底是上面的还是下面的。接下来通过序列比对去比对这两种不同的选择,然后通过序列上错误的比对算出两种选择的概率,根据概率来选到底是哪一个剪切位点具体在这个位置上(图10a)。
然后还有一点是他们发现在比对末端的地方,有时候比对软件会直接把reads比对到基因组上,但那个地方好像还有一个剪切位点。这种情况下我们想确定这个地方是直接比对到基因组上没有剪切位点,还是有一个剪切位点。用类似的想法通过比对算概率,然后看哪种选择的概率更高(图10b)。
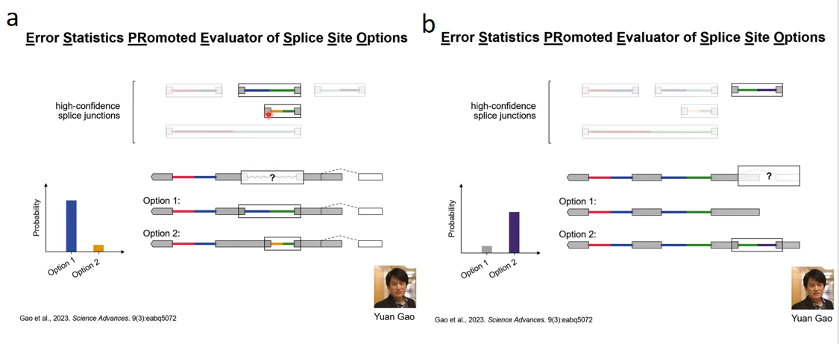
图10. ESPRESSO原理2
这样能得到所有校正后的reads,这些reads中有些是全长的有些不是全长的,我们再把这些reads放到一起去推测全长转录本的序列和表达量。这个问题在20年前已经被解决过,当时第一代转录组测序的技术叫做EST sequencing,这个技术其实也是长读长,只不过当时的通量比现在要差很多,但是很多根据长读长测序做剪切位点或者异构体分析的算法已经建立。所以他们这里就用了当时邢毅教授在做学生时发表的两个算法,一个是集合所有reads得到一个剪切图,然后可以推测全长转录本异构体的序列,另外一个是用EM算法得到全长转录本的表达量(图11a)。
把上述算法集合起来后邢毅教授实验室开发了
ESPRESSO工具,该工具可以从容易出错的长读长RNA测序数据中稳健地发现和量化转录本异构体。这篇文章除了这个算法以外还做了很多其他的事情,用了合成的数据,模拟的数据,还有真实人类RNA的数据比对了各种各样的工具。他们证明ESPRESSO能够很有效的提升转录本的发现和准确定量,尤其是ESPRESSO可以在高置信度下找到新的转录本异构体,而且不需要用同一样本的短读长RNA测序数据做校正。所以这个工作对于长读长RNA测序是一个比较有用的工具,邢毅教授实验室现在很多工作都在用这个ESPRESSO这个工具(图11b)。

图11. ESPRESSO算法
精彩问答
1.现在不管是做癌症还是做细胞系,在做的时候每个样品多少的reads数是比较合适的?
邢毅教授答:他们在ESPRESSO的文章里面有这个分析,他们有一个特别深的illumina的测序数据,然后他们对同样的RNA做了Nanopore测序,再去进行比较,看Nanopore的测序数据在什么样的测序深度下能够得到质量比较好的剪切位点。他们发现至少在那个分析里面到5 million的reads的时候是一个拐点,就是说在5 million的reads之上的话,他们感觉短读长和长读长匹配的还挺好的,但是低于5 million的reads的时候就不行了。
但实际上5 million的long-reads有点凭直觉,因为以前大家都是习惯于做illumina测序,然后就经常听人说得做60 million或者100 million的reads才能得到比较好的结果。实际上你一条long-read不是一条short-read,一条long-read的长度在人类的转录组上面可能就有1 kb或者1.5 kb,所以一条long-read它本身携带的测序信息可能就相当于6-7条short-reads。
如果这样想的话,5 million的long-reads的信息内容相当于大概25-30 million的short-reads的信息内容,已经到了做基因表达量分析觉得还不错的阶段。他们实验室最近的工作在大多数情况下,如果做癌症样本或者细胞系样本,基本上是大概做15-20 million的long-reads。他们觉得做到那个程度的话深度还是挺深的,因为如果把15 million的long-reads再乘以六的话,相当于有90 million的short-reads。
YI XING教授此次会议报告已收录Guangzhou RNA club bilibili视频网站(https://www.bilibili.com/video/BV1gc411g7yu/?spm_id_from=333.999.0.0)
欢迎关注Guangzhou RNA club公众号、网站(rnaclub.rnacentre.org)、twitter(@RNA_club)。
 Guangzhou RNA club
Guangzhou RNA club