Professor Janusz M. Bujnicki, who is a Member of the Polish Academy of Sciences, a Fellow of the European Molecular Biology Organization, an Executive Editor of the journal Nucleic Acids Research, and a Professor at the International Institute of Molecular and Cell Biology in Warsaw, will deliver a lecture titled "Computational modeling of RNA 3D structure and interactions using experimental data" to the Guangzhou RNA Club of the International RNA Society on Thursday (March 16, 2023) at 4pm. Janusz M. Bujnicki's research group uses the SimRNA program to predict RNA structures, and their predicted structures have been ranked among the top performers in the RNA puzzle competition. Their developed approach is used for computing models of RNA's three-dimensional structure and its interactions with other molecules based on these predictions.
Our research focuses on the structure of RNA and its interaction with RNA proteins. Using biophysics and computational simulation techniques, we explore the dynamic behavior and structural features of RNA and develop new RNA structure prediction algorithms. Additionally, we investigate the mechanism of action of RNA proteins, especially those related to diseases. Our research involves a variety of RNAs and RNA proteins, including traditional mRNA and tRNA, as well as non-coding RNAs and RNA proteins that play a key role in diseases.

Figure 1. RNA, the main object of research in the 21st century
Nowadays, not only the scientific community but also the general public have become aware of the importance of RNA and understand its critical role in fundamental biological processes. The threat of RNA viruses, in addition to the well-known SARS-CoV-2, also includes many other viruses that pose a risk to humans and our livestock. At the same time, RNA molecules can also become a type of drug. We have already been able to curb the spread of SARS-CoV-2 through the development of RNA vaccines, which were initially developed to combat cancer. Now, RNA vaccines are likely to become one of our important weapons against many different diseases.
RNA is not only a passive carrier of information for protein synthesis, but it also has many other functions. In fact, RNA is very important in all living organisms, from bacteria to archaea, plants, animals, and humans. Interestingly, RNA can be viewed both as a drug and as a drug target (as shown in Figure 2).
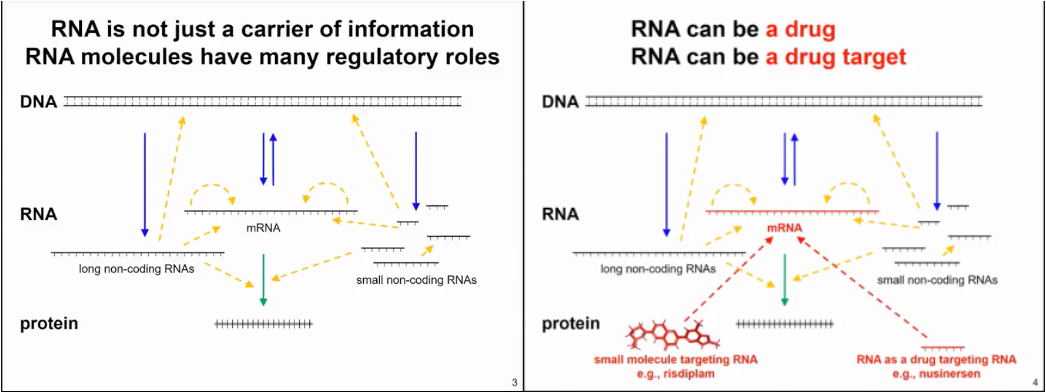
Figure 2. RNA production and function mode
The Janusz M. Bujnicki team is currently researching the issues related to RNA sequences, which all have a common premise - how the three-dimensional structure of RNA is encoded by nucleotide sequences. It is well known that RNA structure is not very stable and can change. The ability of RNA to undergo structural changes is the basis for its molecular function. Therefore, we hope to understand what determines the morphological variability of RNA and how to regulate this variability, for example, through environmental factors, binding of other molecules, or chemical reactions. We also want to understand how RNA can recognize other molecules and how they are recognized by these other molecules. Of course, this process is bidirectional - some RNAs are thought to interact with many molecules, and in reality, many molecules can recognize different RNAs.
That sounds like a very interesting and challenging research focus. The key to understanding the relationship between RNA sequence and structure is to use a combination of computational and experimental methods to fill the gaps in bioinformatics approaches. This involves collecting and organizing existing data and then developing knowledge-based methods for learning and summarizing relevant features from the data. At the same time, it involves predicting unknown parts, such as RNA structures that have not yet been experimentally identified. We then test these predictions in experiments, not necessarily by resolving the structure, but rather through additional data that can confirm or disprove the model. Based on the results, we further validate and improve the methods, integrate the data, and infer general principles.
These efforts are particularly important for the study of RNA molecules because there are relatively few RNA molecules with clearly resolved structures compared to protein molecules. For some RNAs, such as tRNA, there are multiple structures that exist. Many independent or complexed structures of tRNA have been experimentally determined, especially in the ribosome. Therefore, multiple different ribosome and tRNA structures can be found in the PDB database. However, in reality, there are only about 150 to 200 truly different types of RNA whose structures have been determined. The number of available RNA structures is relatively small.
We need more experimental methods to determine the structure of RNA. For example, we can use predicted results to supplement our understanding of RNA structures. Our predictions are based on existing experimental data. Therefore, our modeling method can be considered an effort based on pure theory to discover structural information. In fact, with the theoretical tools available to us, we can make predictions for some structures. For example, we can generate models of RNA 3D structures starting from their sequence. As someone with a background in experimental biology, my favorite way to understand RNA structures is by combining experimental data with computational predictions. We can obtain biochemical and biophysical data for different molecules, but in many cases, the amount of data is insufficient or the resolution is not high enough to directly determine the RNA structure. Of course, if we have high-resolution crystal density maps or very good NMR data, the structure determination process is relatively easy to automate. However, in many cases, the data resolution is relatively low. Therefore, we have density results from molecular density maps or cryo-electron microscopy, but they are difficult to interpret. We can see the rough shape of the molecule, but we do not know the exact position of each nucleotide residue. There are other methods with even lower resolution, such as small-angle X-ray or neutron scattering, which describe the distribution of distances within molecules, but we can only deduce very general structural information (Figure 3).
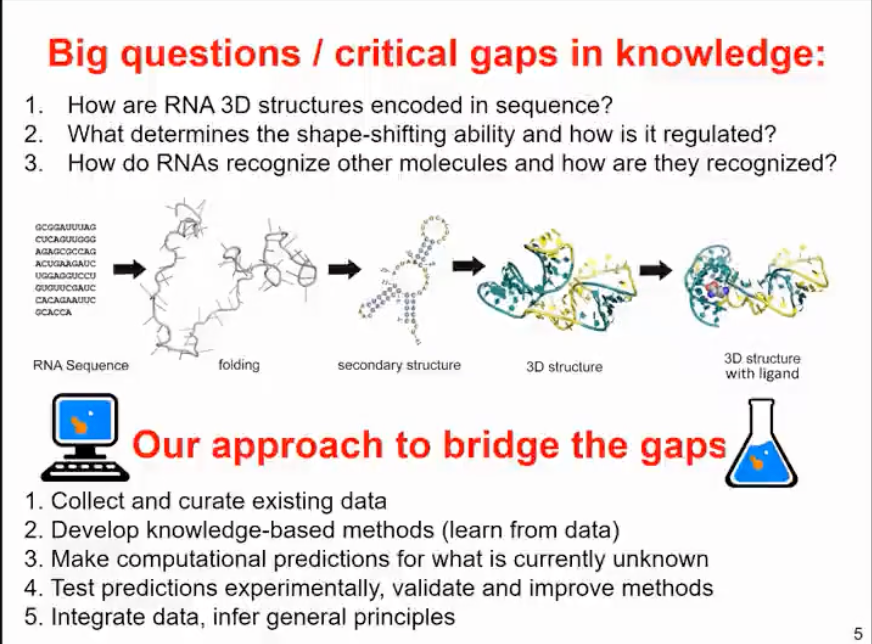
Figure 3. How to solve RNA structure-related problems
There are many chemical probe methods that can be used to analyze the chemical structure of RNA, mainly depending on their structural properties. Chemical probes are often considered as methods for determining RNA secondary structure, but in reality, they only provide chemical information about the structure state of a single molecule. Then, we use computational methods to predict the secondary structure and possible interactions of RNA, and combine experimental data to derive structural models that satisfy different types of experiments.
On the one hand, for computational analysis, we can also use various methods. Starting from first principles, we can use our knowledge of physics to determine the properties of any substance. However, these calculations are very expensive. It is difficult to simulate RNA dynamics using quantum mechanics. On the other hand, we can use databases of known structures and extract information from them. Many machine learning methods rely on this. Our method lies between these two approaches. We try to combine our understanding of the physics and properties of RNA molecules with knowledge-based methods. We have developed simplified representations that can simulate RNA molecule structures and folding relatively quickly. Therefore, we not only focus on predicting RNA structure, which is actually the goal of many machine learning, especially deep learning methods. We also start with some data (such as sequence and other information) and then derive the final structure. For us, it is important to simulate the process of structure formation and how it changes during this process.
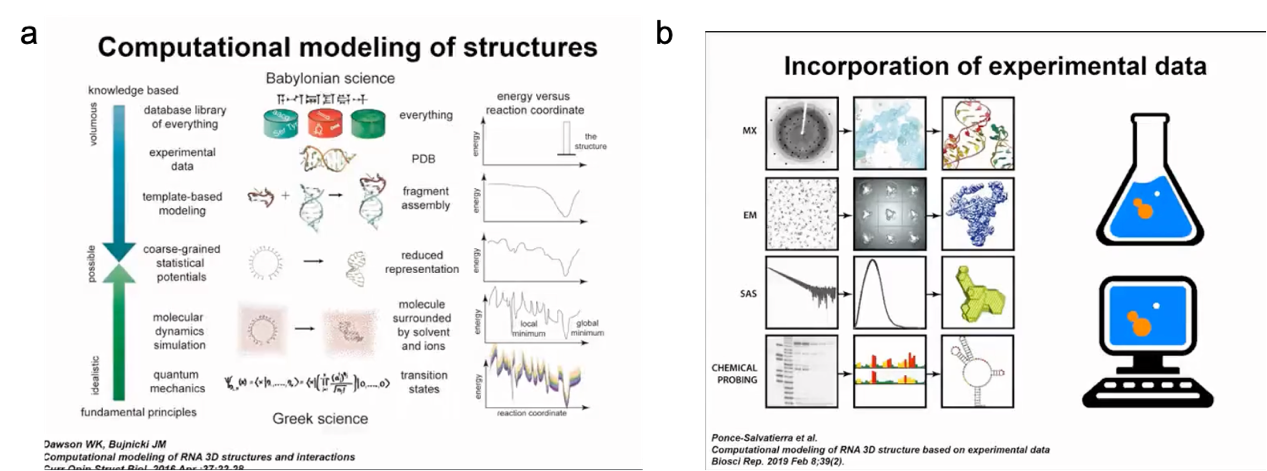
Figure 4. Computational methods for modelling 3D structures in conjunction with experimental data
The method developed in our laboratory is called SimRNA, where "Sim" stands for simulation and simplicity. It uses a coarse-grained representation with three atoms for each nucleotide base to represent purines and pyrimidines, and two atoms (P and C4') to represent the backbone. In simulation, we use the Monte Carlo method to sample the conformational space of the structure and jump between different conformations. This method allows us to simulate the structure and dynamics of RNA molecules quickly and efficiently, giving us a better understanding of RNA folding processes and functions. Since a coarse-grained representation is used, this method also reduces computational costs, making large-scale RNA structure calculations possible.
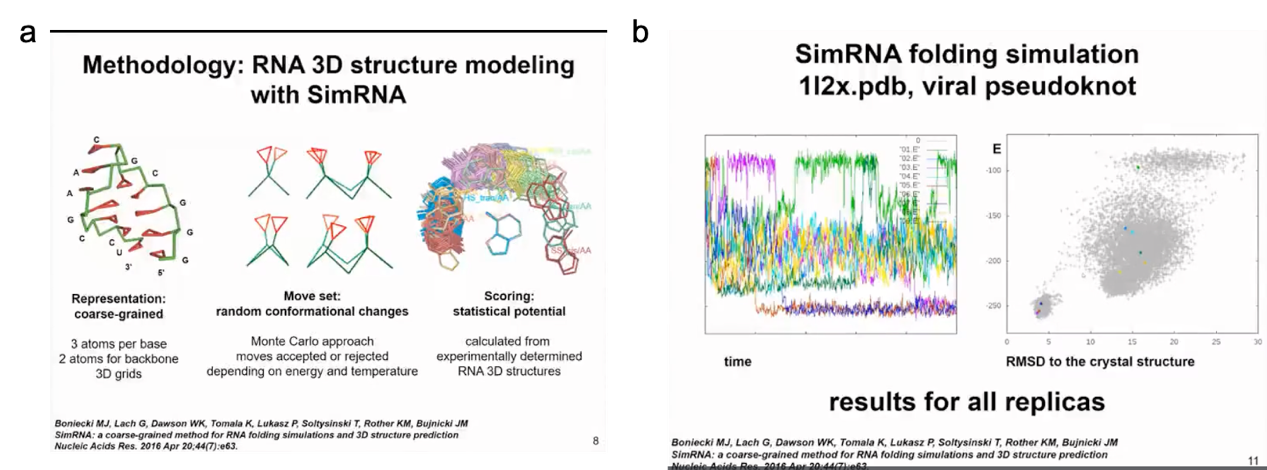
Figure 5. SimRNA program and conformational sampling using MC
When scoring the simulated structure, we use statistical potential energy calculated from experimentally determined RNA 3D structures. For example, in our model, we do not have hydrogen atoms, and therefore we cannot directly calculate hydrogen bonds. However, we calculate the frequency of different orientations of intermolecular interactions and angles within the backbone, as well as the frequency of the target object observed in the existing structure. Thus, by comparing random interactions and volume interactions, we evaluate whether a given state may have lower energy.
I will demonstrate a very simple example of molecule folding. This is a relatively small virus mimic. This simulation will show how SimRNA samples without any additional information. In this simulation, we do not use any constraints, so we do not tell the program where to find suitable structures. In more complex simulations, we can combine the information of the secondary structure we determined. In this simulation, the molecule in the left panel will change its conformation. You will see the molecule moving very quickly to form a compact structure. In the right panel, you will see the relationship between energy and distance from the crystal structure. We do not force the molecule to be close to the actual structure in the crystal, this is only the result of the simulation. Initially, the energy is high. Of course, in the simulation, we hope that the energy decreases and the structure becomes more stable. Now I will run the simulation and hope you can see it. The molecule twists in space, sampling different conformations. The energy was initially high, but now it has become lower. However, the molecule did not completely fold. A helix has formed in the 5' end helix, but the 3' end is still sampling conformations and trying to find a way to fold RNA. Finally, it successfully found the correct pairing mode, reduced the energy, maintained the folded state of the structure, and only sampled adjacent states.
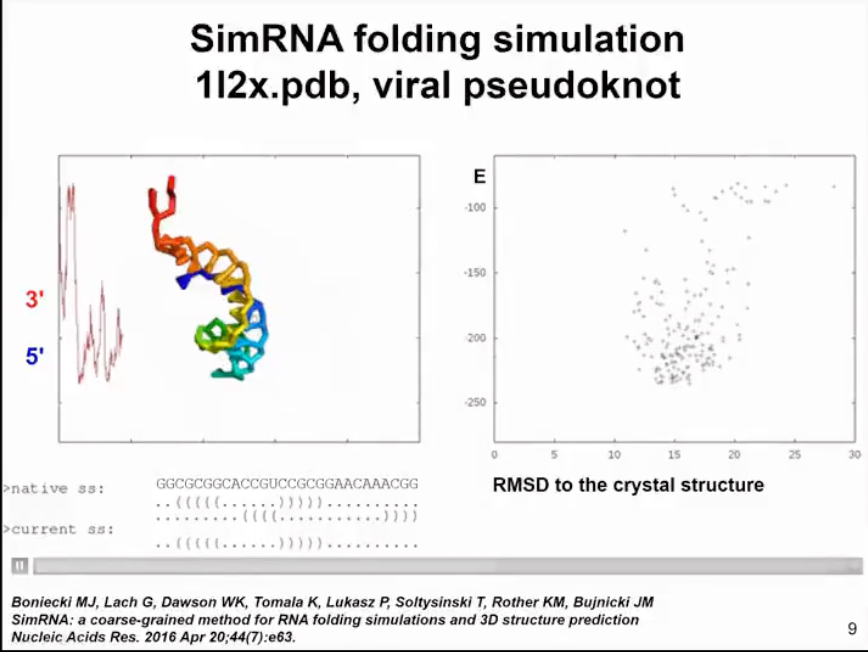
Figure 6. structure and energy change relationships predicted by simRNA
Our method actually uses a technique called Replica Exchange Monte Carlo (REMC) to run simulations at several different temperatures. At higher temperatures, the molecule can partially unfold and sample different conformations, while at lower temperatures, we only minimize its energy. There is a certain probability of exchange between simulations at different temperatures, allowing us to thoroughly sample the conformational space. If the molecule gets trapped in a local energy minimum, we can increase the simulation temperature to jump to another energy minimum. In this molecule, we sample conformations to find the global energy minimum, partially unfolded state, and fully unfolded state. This method not only allows us to obtain the folded structure but also finds all intermediate folding states and identifies the energy barriers between the unfolded and folded state. For more complex cases, we are able to identify additional energy minima (as shown in Figure 7).
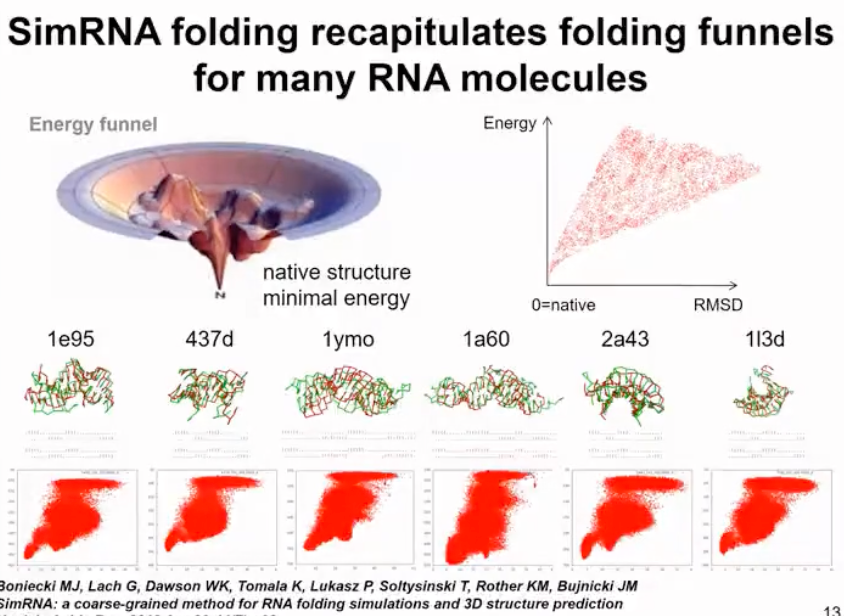
Figure 7. simRNA run results display
However, overall our goal is not to fully reproduce the folding process but to make its energy function smooth so that it is easier to find the minimum values. In reality, energy functions are highly irregular and have multiple energy minima and maxima. That's why physics-based functions can't fully track structures. In our method, we smooth the energy function to quickly overcome energy barriers. It's like sliding on a smooth snow surface during winter when snow is falling on the mountain. Our method works similarly and can find structures for different molecules. We have used similar simulation methods for pseudonucleotides and always found the structure corresponding to the maximum energy minimum. In some cases, we can also find alternative secondary structures. The largest molecule we have analyzed so far is the SARS-CoV-2 RNA genome, which consists of 30,000 nucleotides. Clearly, we didn't fold the entire molecule, which is meaningless since it is highly flexible. But we identified persistent folding regions in the SARS-CoV-2 RNA genome and used them as drug targets.
We initially obtained a complete model of the SARS-CoV-2 RNA secondary structure by analyzing chemical probe data and the conservation of known structural elements. In our analysis, we used chemical probe data obtained in vitro and in cells and found them to be very similar. Although there were some differences, we found multiple highly folded regions in the SARS-CoV-2 RNA not only in untranslated regions but also in translated regions that may have regulatory functions. This was one of the first publications analyzing SARS-CoV-2 RNA, followed by other studies. In our analysis, we used chemical probe data obtained in and out of cells and found very high similarity (as shown in Figure 8).

Figure 8. SARS-COV-2 genome and potential drug targets
Although there are some regional differences, we found multiple regions in the SARS-CoV-2 RNA genome that tend to have high degrees of folding, not only in untranslated regions but also in translated regions that may have regulatory functions. From these various secondary structures, we identified approximately 10 segments throughout the SARS-CoV-2 RNA genome that fold not only in SARS-CoV-2 but also exhibit similarity with other coronaviruses. These are just examples of two small structural regions that are relatively small and form junctions (as shown in Figure 9).
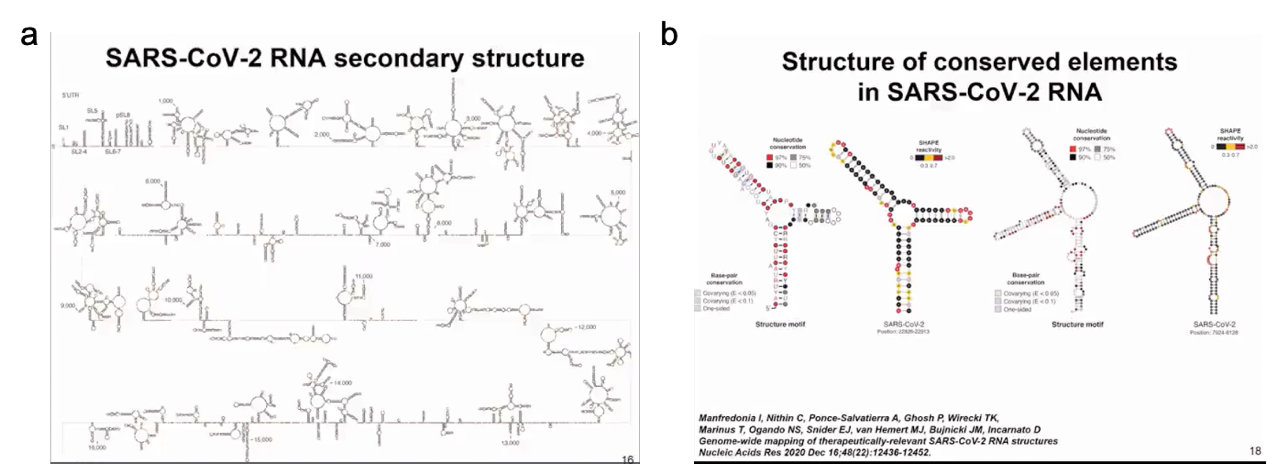
Figure 9 Secondary structure of SARS-CoV-2
We aim to identify sites that can be targeted by small molecule drugs. There have been analyses of RNA and protein structures that show how to identify such binding pockets. Here, we used our simulation method. We sampled the top 1000 structures with the best scores, so it's not just one structure but the entire set of structures, and used the fpocket method to search for sites where small molecules could potentially bind. We haven't yet specified which small molecule might be the target. We're just looking for pockets that could serve as binding targets for small molecule drugs (as shown in Figure 10).
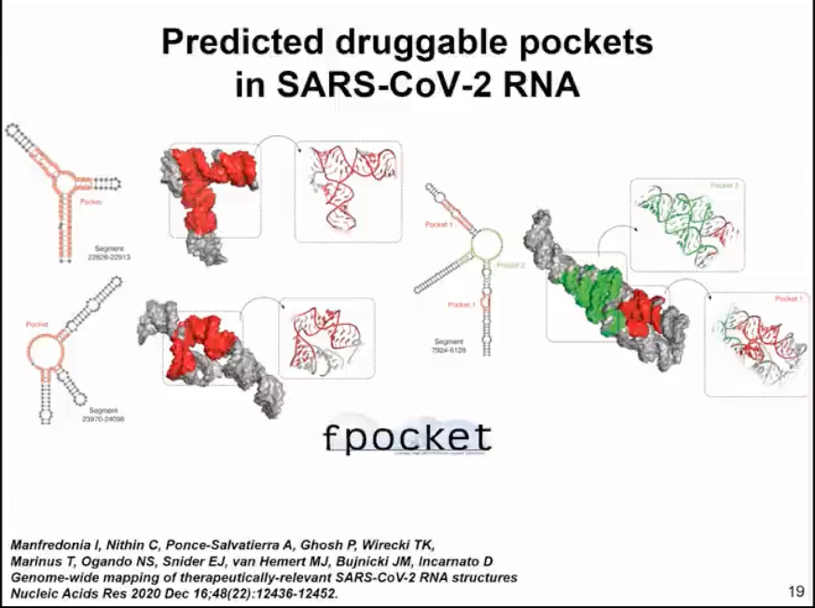
Figure 10. fpocket identification of drug binding pockets
Following this analysis, we also developed some tools that can automatically predict potential folding and drug targeting sites in RNA molecules without the need for chemical probe data. In this published analysis, we did not dock any small molecules because there weren't many available methods. This prompted us to develop a new method, a computational approach for RNA and small molecule docking and scoring. The method we developed is called Annapurna (as shown in Figure 11).
It works similarly to SimRNA. Thus, it uses both coarse-grained representations of RNA molecules and representations of small molecule ligands. Annapurna uses statistical potentials to evaluate the binding of different pharmacophores to different regions of RNA molecules. Therefore, when we use computer docking, we can evaluate the binding of different small molecules quite accurately, better than other methods. According to our benchmark tests, Annapurna can generate 3D models that are close to experimental structures for many scenarios and outperforms other methods available at that time in our benchmark tests. However, this only applies to modeling the interaction between small molecules and rigid RNA molecules. Since we are interested in folding and binding, we turned to studying how RNA is regulated by small molecules.
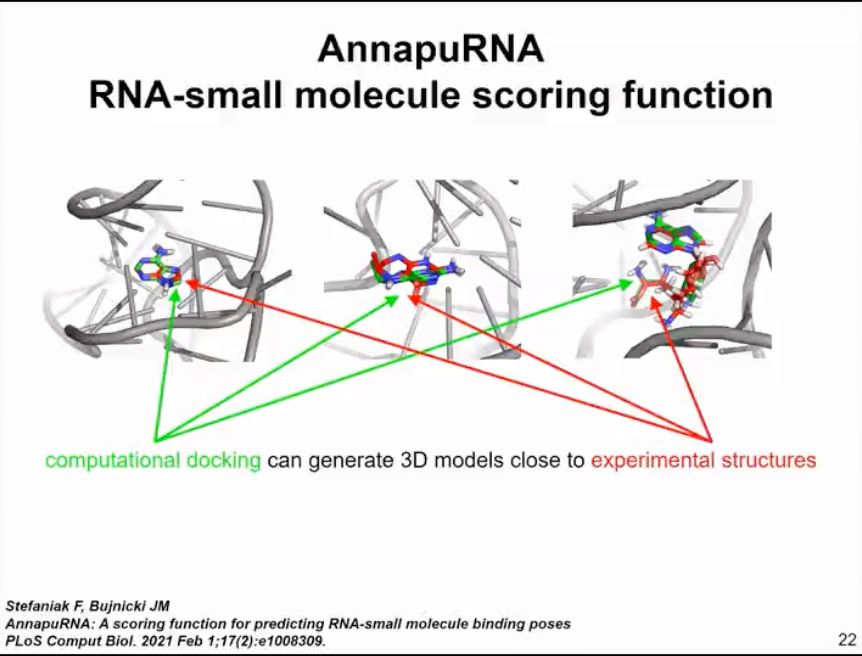
Figure 11. Computational methods for RNA and small molecule docking and scoring - AnnapuRNA
Additionally, when simulating RNA-protein complexes, we modified the previously developed protein folding simulation method and extended it to represent flat surfaces of some amino acid residues and arginine to simulate stacking interactions in proteins and residue pairing with RNA. This idea is similar to the method we used in SimRNA. Therefore, in this work, our real goal is not to predict new protein structures, but to be able to simulate conformational changes within proteins and simulate their interactions with RNA. Therefore, for SimRNP involving RNA and protein interactions (as shown in Figure 12), we usually need additional information about the protein structure. Thus, in most cases, we can actually predict the protein structure quite confidently and can predict the range of dynamics. Therefore, we usually start with a protein that is at least constrained or can be frozen. Then we simulate the binding of RNA or we constraint the RNA and simulate the binding of peptides. Here's an example simulation where we simulated a protein that includes an RNA structural domain and its RNA target by free folding.
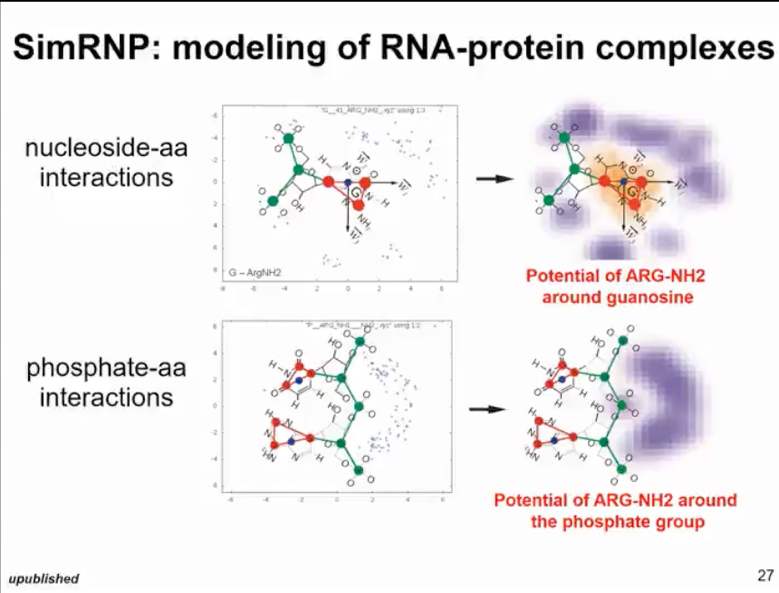
Figure 12. simRNP: simulated RNA and protein complexes
With the SimRNA-cry method (as shown in Figure 13), we can use this approach to handle molecules even if their shape is determined by experiment and the resolution is relatively low. We cannot see the specific details of the helix, only very rough information. We can use simplified simulations to optimize the initial model so that it better fits this situation.
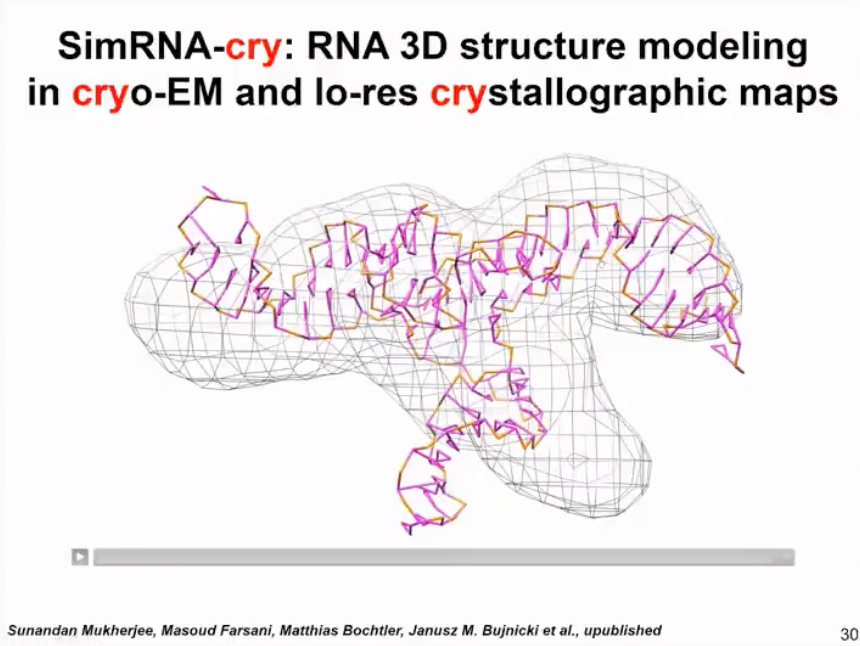
Figure 13. SimRNA-cry results and crystallographic map
We have also developed a method for handling small-angle scattering data, which is similar to SimRNA in assembling structures into compact shapes. This method uses fragments and energy functions, with the fragments coming from our RNA bricks database. We use the information from small-angle scattering data not as a direct shape but as the Poisson ratio of our curves. Thus, we can calculate the difference between the simulated small-angle scattering data of the model and individual models and compare it with experimental small-angle scattering data to find the best fit model for the data. This method can now be used as a web server. This method has not been published yet, but we are preparing for publication.
I must say that in our experience, the resolution of small-angle scattering data is somewhat low in many cases. I advocate using cryo-EM data, which can provide better information about multiple structures and dynamics even at lower resolutions. But sometimes, certain molecules are not suitable for cryo-EM analysis. In such cases, small-angle scattering can serve as a complementary source.

Figure 14. Modelling based on SAXS data
We also have a method for modeling structures based on sparse data and very low-resolution information. Here, we use coarse-graining. But actually, a complete structure can be considered as one particle. Therefore, this method is mainly used to assemble large complexes involving nucleic acids and proteins. We have demonstrated that this can be applicable to very low-resolution experimental data, such as data from negatively stained electron microscopy. We are actually asking a question here, whether the available data is sufficient to generate a model or if the lack of data leads to inadequate results. In fact, we are generating several alternative models that fit the data. Therefore, this method is not only about finding one structure, but answering a question of whether we have enough data, enough experimental data to come to a conclusion about one or multiple structures.
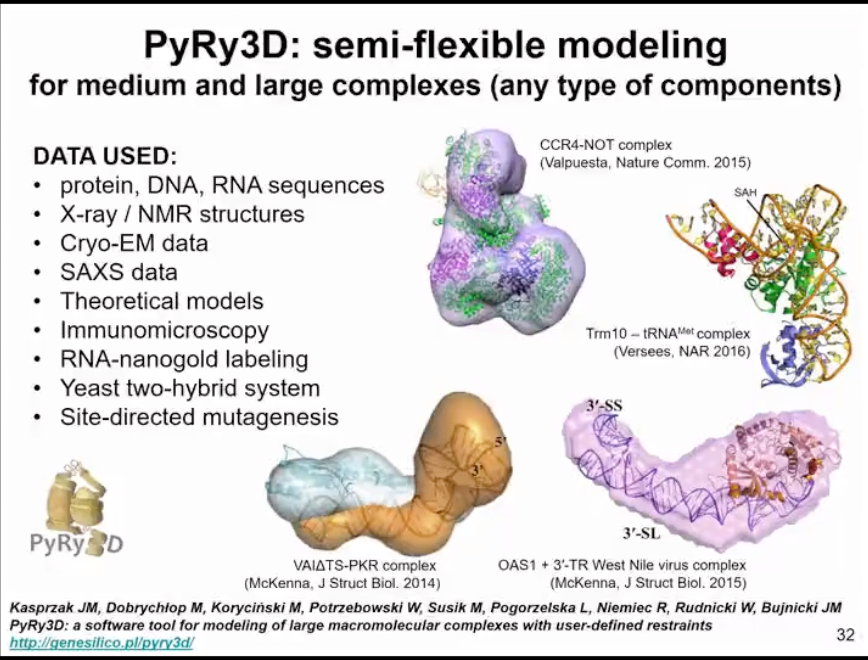
Figure 15. Modelling based on sparse data and low-resolution experimental data
Our current work is to attempt to integrate modeling of different structures in order to model RNA, DNA, proteins, and small molecules simultaneously. Additionally, we're also working on incorporating new types of experimental data into the modeling. As I've mentioned multiple times in this lecture, what we're truly interested in is not just finding one structure, but understanding dynamic processes. Therefore, in this context, we're interested in analyzing and modeling the structures we can obtain to better understand how they impact RNA function, as well as to what extent they are involved in structural transitions. For example, transitions play a crucial role in regulating many processes in your cells. Simulating all of this at once is exceedingly difficult, requiring multiscale simulation including simulating the entire structure within a cell and molecular details. Therefore, we currently have different tools that can solve these types of simulation problems in different stages, and we're now focusing more on molecular details. We don't yet have tools to model these huge complex bodies, but it is definitely the direction in which we're headed.
Q&A part
Now we had know a lot of RNA 3D structures, but what would a valid RNA binding pocket look like? The reason I'm having this question is that even if we have, a small molecule that bound to an RNA binding pocket, how would you expect that we can optimize it? Cuz I mean, one is bound and like, because pretty much the entire surface around it, around the molecule is likely gonna be, maybe mostly hydrophilic and maybe some. So that that will be maybe more like a general question for you.
Matt Disney scripts have actually shown with his research on sort of short molecular. Short fragments of RNA molecules can be targeted by various RNA molecules that do not form extensive hydrophobic interactions. That many of the interactions in groups. It's slightly different than the typical groove in a regular helix. If there's some distortion, for instance, like the insertion of even one. One residue that makes some irregularity in the group can form additional hydrogen bonding structures. As for good urgent donors and acceptors. To distinguish it from the regular helix and make it an attachment point for a small molecule. So some of the groups, like for instance, Matt Disney's, are looking for molecules that can target even relatively shallow pockets. Which can be largely based on hydrogen bonding interactions, and only to a smaller extent by hydrophobic interactions, like, for instance, the intercalation of the aromatic ring.
From the small molecule within the RNA structure. Our approach is slightly different. In our docking, our program anapur now for assessing the poses is capable of finding such molecules. But we found that there that such molecules in docking have relatively low specificity. It is difficult to find very specifically interacting molecules for such shallow pockets. Instead, already in our analysis of Sars-Cov-2, where we modeled the RNAs, we are looking for deeper pockets, especially ones which are formed at junctions and places that have loops. But usually like in the insertion of a loop of several residues only on one side of the helix, Where the helix essentially stays in a stacked format, and then there are additional residues forming and extra interacting elements that can be used by the small molecule.
By our approach, our initial approach was to target very rigid pockets, which is relatively limited. So here we have to know that there would be at least one structural confirmation with a rigid pocket. This will be probably applicable to riboswitches and ribozymes and molecules like that, which can be actually crystallized in the form and in a way where, where, where you where we can visualize the pocket. So we see the crystal or we see the structure from cryo-em and we see a pocket.
So this is a good target for looking for a small molecule that would bind there. However, we are interested in molecules that are also flexible, where we don't have a single structure.So some things could not be really determined using, for instance, crystallography or Quantum.But where from chemical probing data, we see very clearly that there is a preferred local structure formed in a given. Even though the secondary structures move around, we can predict that there would be specific structural arrangements providing a platform to bind the small molecule.
In this case, the goal of the binding of the small molecule would be to target one of the many forms that exist in given RNA. And to be able to shift the confirmation equilibrium towards one specific confirmation, thereby having the effect.
Professor Janusz M. BujnickiThe report of this meeting has been included in the Guangzhou RNA club bilibili video website:(https://www.bilibili.com/video/BV1vx4y1P7zc/?vd_source= f97b88c9400c9d0b88e6bbfbb3ff8294)
Welcome to follow Guangzhou RNA club on public website (rnaclub.rnacentre.org) and twitter (@RNA_club).
References
Boniecki, Michal J et al. “SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction.” Nucleic acids research vol. 44,7 (2016): e63. doi:10.1093/nar/gkv1479
Rother, Magdalena et al. “ModeRNA: a tool for comparative modeling of RNA 3D structure.” Nucleic acids research vol. 39,10 (2011): 4007-22. doi:10.1093/nar/gkq132
Boccaletto, Pietro et al. “MODOMICS: a database of RNA modification pathways. 2017 update.” Nucleic acids research vol. 46,D1 (2018): D303-D307. doi:10.1093/nar/gkx1030
Cappannini, Andrea et al. “NACDDB: Nucleic Acid Circular Dichroism Database.” Nucleic acids research vol. 51,D1 (2023): D226-D231. doi:10.1093/nar/gkac829
Pawlowski, Marcin et al. “MetaMQAP: a meta-server for the quality assessment of protein models.” BMC bioinformatics vol. 9 403. 29 Sep. 2008, doi:10.1186/1471-2105-9-403
Tuszynska, Irina et al. “NPDock: a web server for protein-nucleic acid docking.” Nucleic acids research vol. 43,W1 (2015): W425-30. doi:10.1093/nar/gkv493
Magnus, Marcin et al. “SimRNAweb: a web server for RNA 3D structure modeling with optional restraints.” Nucleic acids research vol. 44,W1 (2016): W315-9. doi:10.1093/nar/gkw279
 Guangzhou RNA club
Guangzhou RNA club